从最新的JEV质量评价框架,看外泌体检测如何从“基础表征”走向“产品质控”
随着国务院令第818号(简称818号令)《生物医学新技术临床研究和临床转化应用管理条例》于2025年9月28日正式颁布,到今年4月份,恩泽康泰合作伙伴上海思德克索的IND申请被国家药品监督管理局药品审评中心(CDE)受理,再到今年5月1号818号令的正式实施,细胞外囊泡(EVs)已经从科研热点走向了深度的治疗与应用场景。尤其是干细胞来源EVs,在组织修复、免疫调节、再生医学和递送系统中持续升温之后,行业真正关心的问题已经发生变化:不只是“有没有外泌体”,而是“这批外泌体是否来源清楚、质量稳定、功能可证、安全可控、能够用于后续开发”。
近日,由中国食品药品检定研究院、国家药品监督管理局药品评价中心、药品监管科学国家重点实验室以及北京恩泽康泰(Echo Biotech)等单位共同完成的文章《Quality Evaluation Considerations for Stem Cell-Derived Extracellular Vesicles-Based Therapeutic Products in China》正式在JEV上见刊,首次系统地提出了干细胞来源EV治疗产品的质量评价思路。把EV产品的开发从“研究表征逻辑”真正推向了“药学质量评价“的逻辑。
以前大家能参考的MISEV2023等指南回答的是基础研究中“如何证明研究对象是EV、如何保证实验可重复”等问题;而这篇文章进一步面向治疗产品开发,强调细胞来源、身份鉴定、理化属性、强度、外源污染、纯度、生物学功能、安全性、稳定性、制剂和方法学选择等关键质量属性(CQAs)。这对外泌体的质量检测也提出了更清晰的方向:检测不能只是拍图、跑条带、测粒径,而要围绕产品质量属性建立一套可解释、可比较、可复用的检测矩阵。
下面,我们就逐项深度剖析文章中的质量评价逻辑,结合外泌体质量检测分析一站式解决方案,逐项拆解每类检测:检测什么、原理是什么、结果回答什么问题,有哪些局限性,以及结果解释中常见的争议点等。
一、来源与身份鉴定:首要回答“它是谁、从哪里来”
文章首先强调,干细胞应被视为EV生产的核心原材料。EV的货物组成和生物学功能会反映来源细胞状态,因此细胞来源、培养体系、传代稳定性和细胞库质量都会影响最终EV产品。落到检测端,第一步就是建立EV身份鉴定和来源相关标志物评价。
WB(三阳一阴)是最常见的身份鉴定方法,通常检测CD9、CD63、CD81、TSG101、Alix、HSP70等EV阳性标志物,并用Calnexin等阴性标志物排除细胞器污染。它的原理是基于蛋白免疫印迹识别样本中的特征蛋白,适合科研表征、样本初筛和论文支撑。争议点在于,WB是整体蛋白水平检测,无法说明每一个颗粒是否携带标志物,也不能充分区分不同细胞来源的EV亚群。
因此,文章提出的“身份”评价,对于人源EV样本,可以进一步采用荧光抗体染色+NanoFCM检测CD9/CD63/CD81阳性颗粒占比,在单颗粒层面观察标志物阳性比例。这类检测更适合批间一致性、工艺优化和企业端质量研究,因为它回答的是“多少颗粒具备目标表型”,而不是“总样本里有没有这个蛋白”。
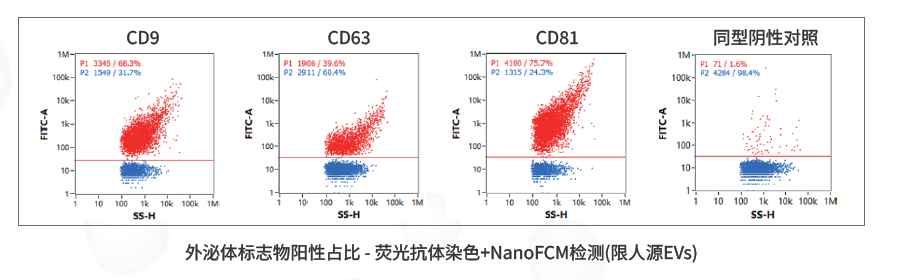
外泌体标志物阳性占比:荧光抗体染色+NanoFCM可在颗粒级别观察CD9/CD63/CD81等标志物表达比例。
对于MSC来源EVs,不能只停留在通用EV标志物,还可以检测CD73/CD90等干细胞相关标志物阳性颗粒占比。它不能替代细胞库检测,也不能单独证明疗效,但可以作为来源相关身份的补充证据,帮助判断EV颗粒是否保留来源细胞相关表型。争议点主要来自抗体特异性、表位可及性、不同来源MSC差异以及仪器阈值设定,因此更建议在同一产品体系内建立内部趋势和合格范围。
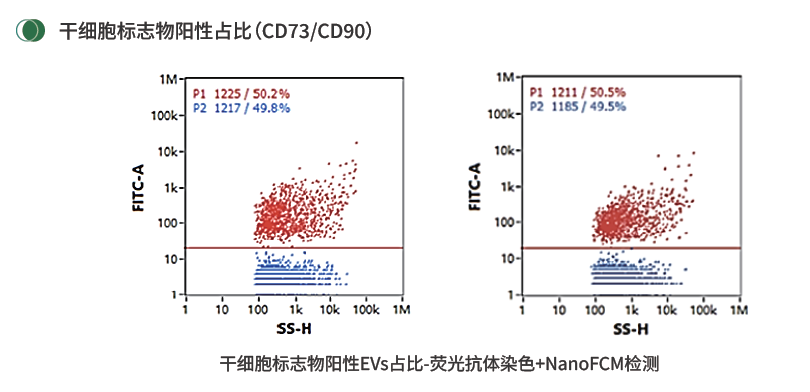
干细胞标志物阳性EVs占比:CD73/CD90等来源相关marker可用于补充MSC来源EVs颗粒级身份评价。
二、理化性质:再回答“它长什么样、分布是否稳定”
文章将EV理化性质作为关键质量评价维度之一,包括形态、粒径分布、颗粒浓度、Zeta电位等。这一部分对应的是外泌体基础表征,但基础表征并不等于“低阶检测”。当它用于工艺比较和批间一致性时,数据质量和方法选择非常重要。
TEM透射电镜用于观察囊泡形貌和膜性结构。经典EV图像常呈“cup-shaped”,但这与负染、固定、脱水等制样过程密切相关,不应被机械理解为天然状态。TEM适合作为形态学证据和异常大颗粒、碎片的直观观察,但由于视野有限、样本代表性不足,不适合单独做定量。
粒径及颗粒浓度可通过NanoFCM、NTA和RPS等方法检测。NTA基于布朗运动轨迹推算粒径,应用广、可视化强,但易受折射率、浓度和杂质颗粒影响;RPS基于颗粒通过纳米孔产生的电阻脉冲,粒径和颗粒浓度定量能力较强,常被用于更严肃的工艺和申报型场景,但对样本过滤、堵孔和缓冲体系更敏感;NanoFCM可在纳米尺度上结合散射和荧光信号分析,是后续阳性颗粒占比、有膜颗粒、膜完整性等检测的基础平台。DLS适合快速观察整体粒径趋势和聚集状态,但它是强度加权方法,少量大颗粒或聚集物会显著影响结果,因此更适合作为补充稳定性观察,而不宜单独用于复杂EV样本定量。Zeta电位通过激光多普勒电泳法反映颗粒表面电荷和胶体稳定性,受pH、离子强度、辅料和缓冲液影响明显。它虽不能直接代表疗效,但对制剂稳定性、聚集风险和运输条件优化很有价值。
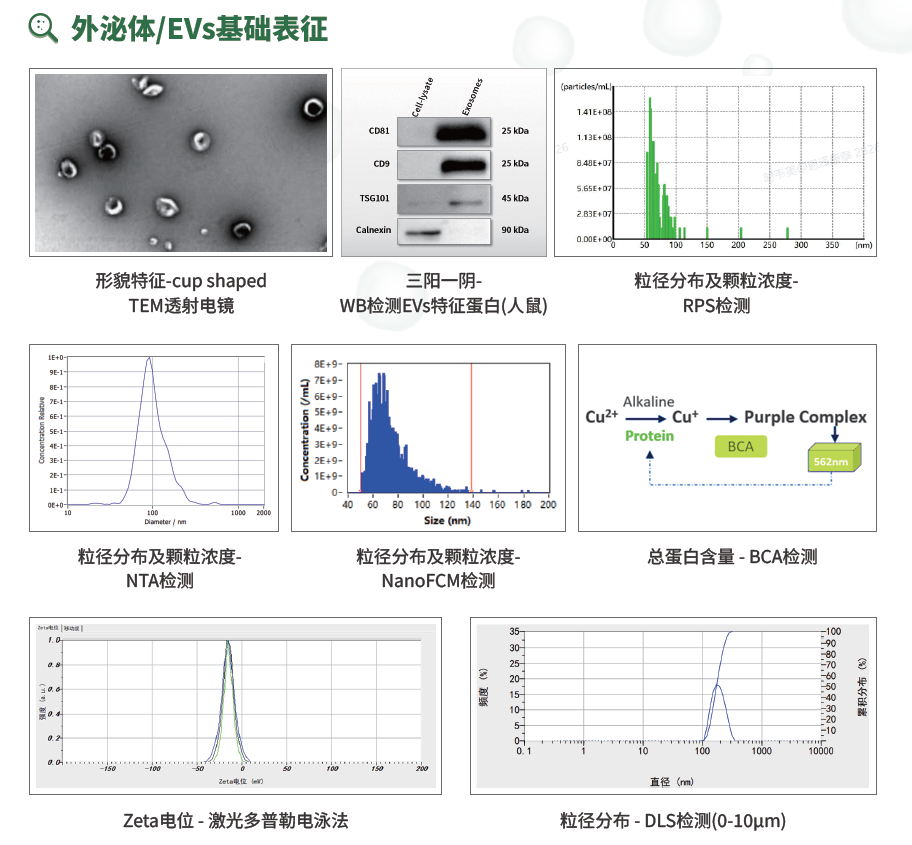
外泌体/EVs基础表征:TEM、WB、RPS、NTA、NanoFCM、BCA、Zeta电位和DLS等方法共同构成基础质量画像。
三、强度与剂量:不能只看“总颗粒数”
文章特别提出,EV产品的强度(strength)不应简单等同于总颗粒数、总蛋白或总RNA。更合理的思路,是在定义数量的EV颗粒中,进一步评价与功能相关的活性成分、阳性颗粒比例或功能效价。
颗粒浓度检测是剂量设计的基础,但它并不天然等于有效剂量。因为检测到的颗粒中可能包含非膜性颗粒、蛋白聚集体、脂蛋白或辅料颗粒。BCA总蛋白可以辅助计算颗粒/蛋白比,帮助判断同一工艺体系下的蛋白污染趋势和分装一致性。但颗粒/蛋白比存在平台差异和来源差异,不建议机械套用为绝对纯度标准,更适合做内部批间比较。
对于工程化或功能型EV产品,活性成分检测可以进一步向标签阳性颗粒占比、多组学分析、候选miRNA/蛋白靶向检测和功能实验延伸。比如,目标蛋白、靶向肽或膜展示蛋白带有FLAG/HA等标签,可以通过目的蛋白标签阳性颗粒占比评价装载或展示效率。这里的关键不是证明细胞表达了目标蛋白,而是证明目标信号真正出现在EV颗粒上,能有效体现出有多少EV颗粒携带了目的蛋白。常见的争议包括游离蛋白污染、蛋白聚集体误判、表位是否朝外暴露、抗体是否可识别等,因此必须设置同型阴性对照和合适的颗粒门控策略。另外,多组学适合研发阶段发现候选CQA,但不适合直接作为常规放行检测;更靠谱的路径是先通过多组学发现候选活性分子,再通过qPCR、ELISA、靶向蛋白检测或体外效价模型建立可重复的质量指标。
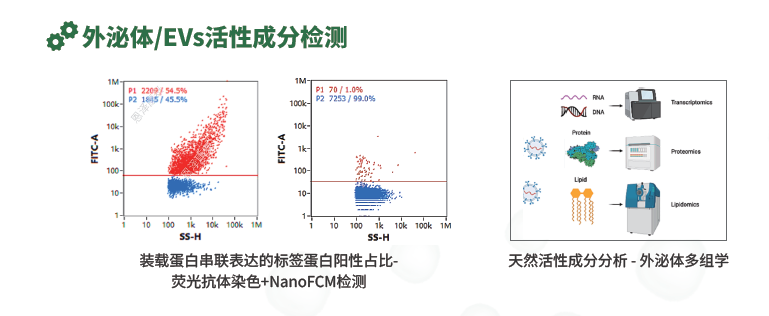
外泌体/EVs活性成分检测:工程化标签阳性颗粒占比可用于验证装载效率,多组学更适合发现候选活性成分。
四、外源污染:治疗性EV必须排除底线风险
文章在外源污染部分强调,EV生产和制剂过程中可能引入细菌、真菌、支原体、内毒素、病毒、外源蛋白、外源核酸或工艺残留。对于科研样本,这些项目常被忽视;但一旦进入动物实验、临床前研究、注射制剂或企业端放行,外源污染就是底线问题。
无菌检查包括直接接种法和快检。直接接种法按照药典思路培养周期更长,可覆盖需氧菌和厌氧菌,更适合严肃安全性评价;快检速度更快,但覆盖范围有限。样本如含抑菌或杀菌成分,需要提前说明,否则可能出现假阴性。
支原体检测常用于细胞培养体系、细胞上清和EV样本风险排查。支原体污染会改变细胞状态、影响EV组成,也可能干扰免疫相关功能实验。内毒素检测通常采用凝胶法等药典方法,但EV样本中的缓冲液、辅料、蛋白和复杂基质可能造成干扰,因此需要提前预估内毒素范围并进行方法适用性判断。
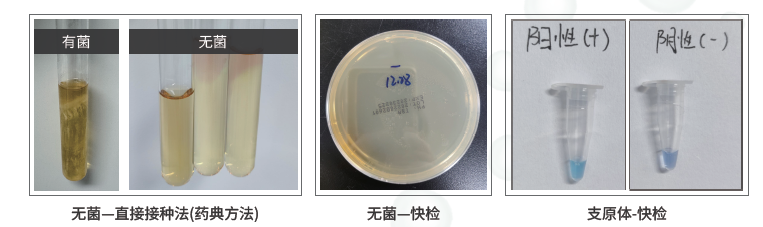
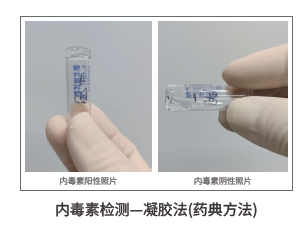
外泌体/EVs药典推荐及安全性检测:无菌、支原体、内毒素排除底线风险
五、纯度:真正重要的是“有效颗粒占比”
文章将纯度列为EV治疗产品质量评价的关键维度。纯度不是一个单一指标,而是由多个层面共同构成:目标粒径范围内颗粒比例、膜完整颗粒比例、携带标志物或活性成分颗粒比例、蛋白/核酸污染、非目标细胞来源EV、工艺残留和辅料干扰。
有膜颗粒占比可通过Triton裂解+NanoFCM评价。Triton破坏脂质膜后,裂解前后颗粒变化可用于推测膜性颗粒比例。它适合区分膜性颗粒与部分非膜性颗粒,但某些脂蛋白或非目标膜结构也可能受到影响,样本杂质复杂时波动会增大。
膜完整性可通过Calcein-AM染色+NanoFCM评价。该方法关注具有完整膜结构并产生荧光信号的颗粒比例,尤其适合冻融、运输、储存、制剂辅料和稳定性研究。争议点在于血清、酚红、外源酯酶、未知缓冲体系等可能干扰反应,因此样本体系必须清楚,必要时需要换液或设置空白对照。
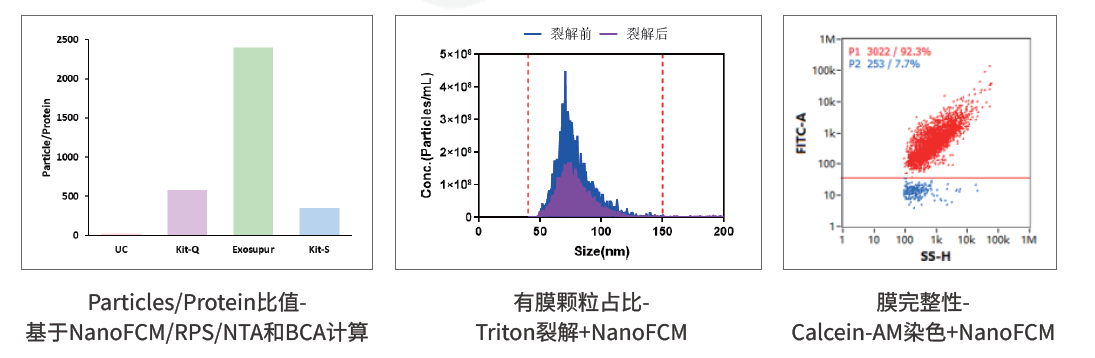
外泌体/EVs纯度表征:颗粒/蛋白比、有膜颗粒占比和膜完整性分别从不同维度回答“有效颗粒”问题。
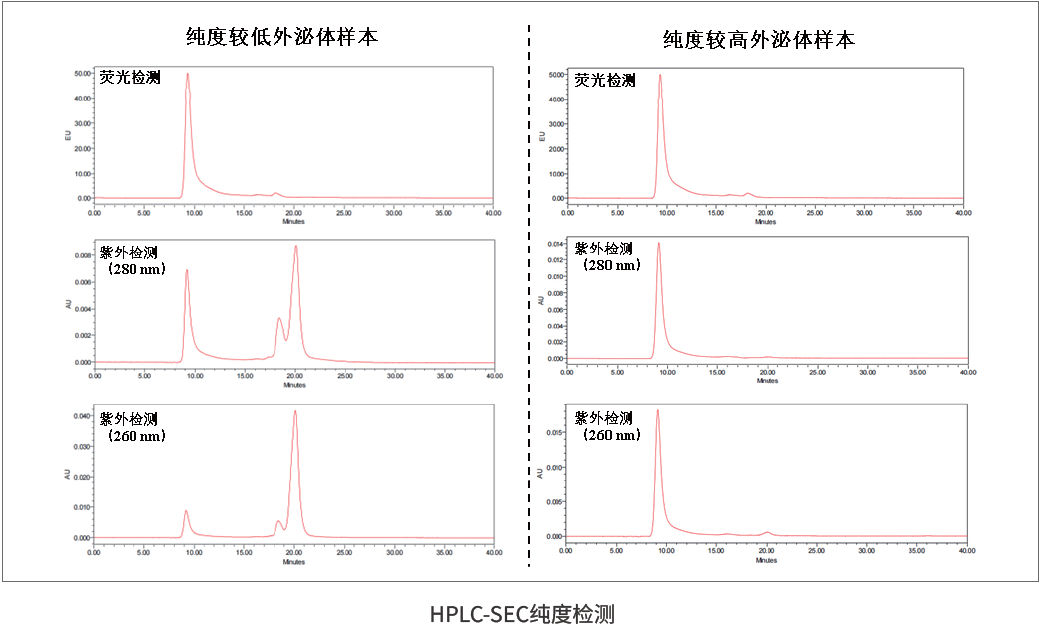
HPLC-SEC纯度检测则从分离和杂质谱角度补充评价。它采用分子排阻色谱思路,可同时观察EV荧光图谱以及260 nm/280 nm紫外信号提示的核酸、蛋白等杂质分布。它特别适合比较不同分离纯化工艺,例如超速离心、试剂盒、SEC柱、TFF浓缩等方案。需要注意的是,HPLC-SEC也不是绝对纯度证明,不同EV亚群、脂蛋白和蛋白复合体可能存在洗脱重叠,因此更适合与NanoFCM、BCA、膜完整性等指标联合解读。
HPLC-SEC纯度检测:通过荧光检测和260/280 nm紫外信号观察EV主峰及蛋白/核酸杂质分布。
六、生物学功能:从“像EV”推进到“有预期活性”
文章提出,生物学功能评价应与产品拟定适应症和作用机制相关。对于干细胞来源EVs,常见功能包括免疫调节、抗炎、促迁移、促修复、促血管生成和抗凋亡等。检测端不能只证明样本“像EV”,还要逐步建立与预期功能相关的体外活性模型。
抗炎活性检测通常采用LPS诱导RAW264.7巨噬细胞炎症模型,加入供试EV后检测IL-6、IL-10等炎症相关因子的变化。该模型适合免疫调节和抗炎方向EV样本。关键争议点在于,样本内毒素、无菌状态和颗粒剂量归一化会直接影响结果,因此建议在功能实验前完成颗粒浓度、无菌和内毒素评估。
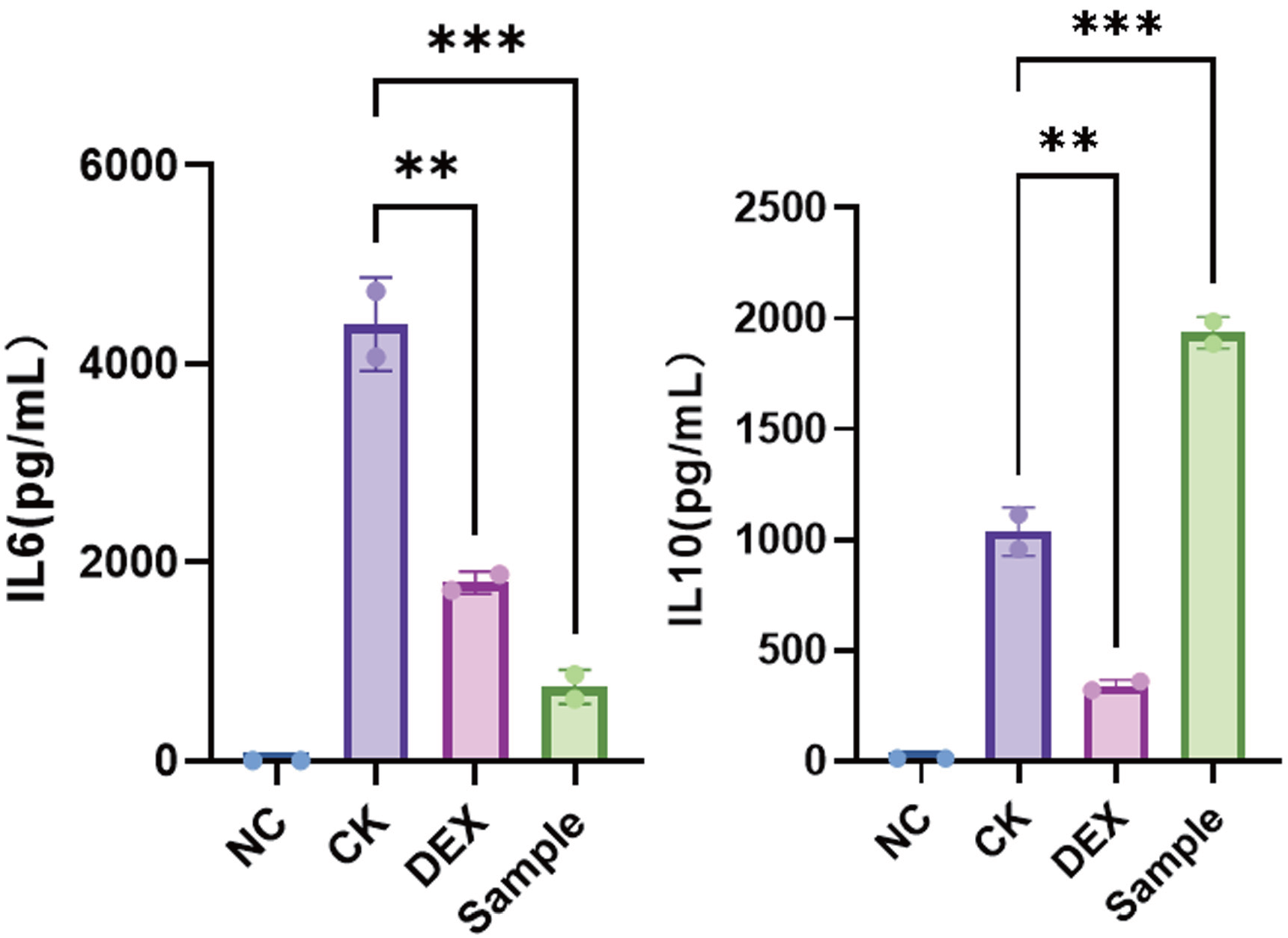
抗炎活性检测:LPS诱导RAW264.7巨噬细胞炎症模型,通过IL-6、IL-10等指标评价EVs对炎症反应的调节作用。
促修复活性检测通常采用成纤维细胞划痕实验,观察EV对细胞迁移和伤口闭合的影响。它适合皮肤修复、组织修复和再生医学方向样本的早期功能筛选。争议点在于划痕实验同时受迁移、增殖、细胞状态、拍照区域和分析阈值影响,因此必须设置未处理组、阳性对照组,并统一EV剂量归一化方式。

促修复活性检测:以成纤维细胞划痕模型评价EVs促进细胞迁移和体外修复的能力。
七、安全性与制剂稳定性:产品开发阶段必须补上的检测
文章在安全性部分重点讨论成瘤性/致瘤性、凝血/溶血风险、免疫原性、残留物以及不同细胞来源EV的特殊风险。检测服务中与常规制剂和药典安全性直接相关的项目,包括无菌、支原体、内毒素、不溶性微粒、pH、渗透压、冻干粉水分等。
不溶性微粒检测关注的是微米级颗粒,与NTA/NanoFCM检测的纳米EV不是同一尺度。对于注射产品,大粒径微粒可能带来血管栓塞、局部炎症和肉芽肿风险。pH和渗透压直接影响给药刺激性、细胞兼容性和制剂稳定性。冻干粉水分检测则用于评估冻干工艺和长期储存风险,水分过高可能加速结构破坏、蛋白/RNA降解或活性下降。
制剂稳定性不是单一时间点检测,而应结合储存温度、冻融次数、缓冲体系、辅料、包装材料和运输条件进行动态观察。建议将粒径分布、颗粒浓度、Zeta电位、膜完整性、HPLC-SEC杂质谱和体外功能活性组合起来,构建稳定性评价矩阵。


外泌体/EVs药典推荐及安全性检测:无pH、渗透压、不溶性微粒和冻干粉水分等项目共同支撑制剂稳定性。
八、方法学选择:研发阶段“看得全”,放行阶段“测得稳”
文章最后强调,检测方法选择需要区分研发阶段和放行阶段。研发阶段适合采用多平台、多维度、正交验证的方法,尽可能识别EV的组成、异质性和潜在功能机制;而放行阶段更强调方法稳健、结果可重复、操作可标准化、与质量决策直接相关。
这也解释了为什么同一个项目在不同阶段需要不同检测组合。早期科研阶段,TEM、WB、NTA/NanoFCM加BCA可能足够支撑基础表征;工艺优化阶段,需要加入HPLC-SEC、有膜颗粒、膜完整性和标志物阳性颗粒占比;制剂和企业端质量研究阶段,则必须补充无菌、支原体、内毒素、不溶性微粒、pH、渗透压和稳定性相关检测;功能型产品还应逐步建立体外活性或效价模型。
恩博推荐检测套餐
1. 科研基础表征套餐:TEM + WB(三阳一阴) + NanoFCM/NTA/RPS粒径及颗粒浓度 + BCA+ZETA电位。适用于论文发表、课题结题、早期样本确认,重点回答“是否具备EV基本特征”。
2. 工艺优化与纯度评价套餐:颗粒浓度 + BCA + 颗粒/蛋白比 + HPLC-SEC + 有膜颗粒占比 + 膜完整性+ CD9/CD63/CD81阳性颗粒占比。适用于比较不同分离纯化工艺、评估杂质背景、筛选更稳定的EV制备路线。
3. 干细胞来源EV身份增强套餐:基础表征 + CD9/CD63/CD81阳性颗粒占比 + CD73/CD90阳性颗粒占比。适用于MSC来源EVs、干细胞分泌组产品和批间一致性研究。
4. 工程化EV验证套餐:基础表征 + CD9/CD63/CD81阳性颗粒占比+ FLAG/HA目的蛋白标签阳性颗粒占比 + HPLC-SEC纯度检测+ZETA电位。适用于靶向肽修饰、膜蛋白展示、递送型EV和工程化EV构建验证。
5. 制剂与安全性套餐:无菌检查 + 支原体 + 内毒素 + 不溶性微粒 + pH + 渗透压 + Zeta电位;冻干制剂建议增加水分检测。适用于动物实验、临床前制剂、注射给药样本和企业端质量放行前评估。
6. 体外活性评价套餐:颗粒浓度归一化 + 无菌/内毒素确认 + 抗炎活性或促修复活性检测。适用于功能型EV产品筛选、批间活性比较和候选效价方法建立。
总结
从CQA建立的角度看,外泌体质量检测不应被理解为若干单项检测的简单拼接,而应是一套围绕“结构-组成-功能-安全-稳定性”关系建立的证据链。TEM、WB、NTA、RPS、NanoFCM、BCA、Zeta、HPLC-SEC、膜完整性、阳性颗粒占比、药典安全性和体外功能模型,分别从不同维度刻画EV样本的身份、异质性、有效颗粒比例、杂质谱、剂量基础、制剂适配性和生物学效应。
真正有价值的EV质控体系,应当做到三个层面的闭环:第一,分析方法学闭环,即关键指标尽量采用正交方法验证,避免单一平台偏差;第二,工艺质量闭环,即同一产品在不同批次、不同储存条件和不同制剂状态下可比较;第三,功能关联闭环,即候选活性成分、阳性颗粒比例和体外效价模型逐步建立相关性。只有当这些数据能共同解释产品的一致性、安全性和预期功能时,外泌体检测才真正从“样本鉴定”进入“产品质量评价”。
恩泽康泰将持续围绕外泌体/EVs基础表征、纯度检测、药典&安全性、活性成分检测、干性marker和体外功能活性,提供贴近产品开发逻辑的一站式质量检测分析服务。
截止目前,一站式的外泌体质量检测分析平台已经助力合作伙伴上海思德克索的IND申请被国家药品监督管理局药品审评中心受理,全面的外泌体检测已经服务超过300家科研机构,检测服务超百家公司,未来将帮助更多的科研和企业客户把EV样本逐步做成可评价、可优化、可比较、可转化的产品,辅助更多的外泌体产品和研究获批上线!